James Bradford
Head of Bioinformatics, Almac Diagnostic Services


Gene expression signatures – Promise vs Reality
Since the advent almost 25 years ago of techniques that enable simultaneous measurement of gene expression in a single sample, the use of gene expression profile combinations (or “signatures”) to understand tumour biology has promised to revolutionise our approach to diagnosis and prognosis in the clinic. However, this potential has yet to be fully realised despite the exponential increase in genomic data during this period. This blog explores some of the issues that have hindered progress, and highlights several recent trends and innovations that may yet fuel a rise in successful translation of gene signatures into clinical application.
Gene expression signature discovery: a history of potential
A gene expression signature refers to a finite, pre-determined group of genes whose combined expression profile is highly specific to a biological process, disease state or pathogenic medical condition. Typically, the signature generation process takes place in three main phases: discovery, development and independent validation.
Discovery: During discovery, gene expression profiles are determined in a set of “training” samples, where genes highly correlated to or differentially expressed between the phenotype(s) of interest (e.g.prognosis) are selected. The training set must be of sufficient size to allow statistically meaningful associations between genes and phenotype to be identified as an underpowered analysis can lead to failure during the independent validation phase.
Development: During development, genes are further refined, and candidate signatures undergo rigorous cross-validation before a method to score and classify patients based on their signature profile is implemented. The discovery and development phases are often indistinguishable, particularly if automated methods such as machine learning are employed.
Independent validation: For translation into the clinic, an independent validation phase, where signatures are tested in clinically relevant cohorts distinct from those used to develop the classifier, is critical.
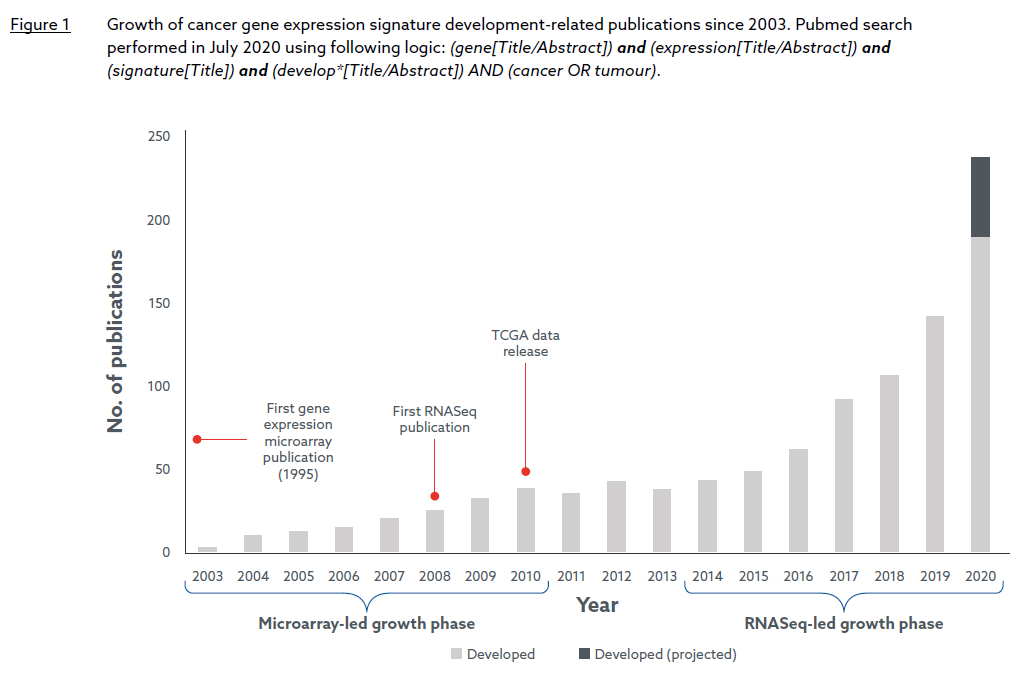
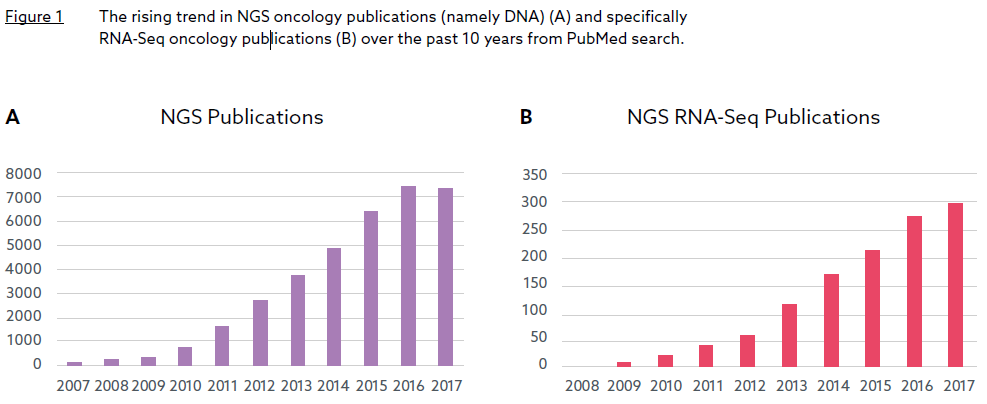
Whilst the first gene expression signature can be traced back to 1995, a simple search of Pubmed reveals that cancer gene expression signature development activities did not become widespread until the early-mid 2000s (Figure 1).

“Currently, over 100 cancer gene signature development efforts are published every year, and in 2020 the number is likely to reach nearly 250.”
Figure 1 suggests two periods of exponential growth (2003-2010 and 2014-present), each catalysed by a major advancement in gene expression profiling technology around five years previous: the cDNA microarray in the mid-1990s and RNA Sequencing (RNA-Seq), its Next Generation Sequencing-based successor, in the mid-2000s. The consolidatory period between 2011 and 2013 appears to coincide with a transition between the two platforms. It is also worth noting that the release of The Cancer Genome Project (TCGA) expression data, generated by both microarray and RNASeq platforms, began in 2010, and has likely provided further impetus for signature development during the last decade. Currently, over 100 cancer gene signature development efforts are published every year, and in 2020 the number is likely to reach nearly 250.
Translation to the clinic: a potential unfulfilled…yet
Despite the opportunities offered by new technologies, and extensive efforts by the MAQC (Microarray Quality Control) consortium1 to show both microarray and RNASeq platforms are sufficiently reliable for clinical and regulatory purposes (at least if mRNA is extracted from high quality samples), the vast majority of signatures have failed to make any clinical impact. Indeed, to the author’s knowledge, only two gene expression signatures have gained FDA approval, both of which are prognostic in breast cancer: Prosigna (a 50-gene signature providing a risk-of-recurrence score) from Veracyte and Mammaprint (a 70-gene signature to stratify patients into high versus low risk for relapse) from Agendia. No gene expression signature has gained approval since Mammaprint in 2013.
Problems associated with the first exponential growth phase of signature development were documented in two seminal papers published just after that period2, 3.
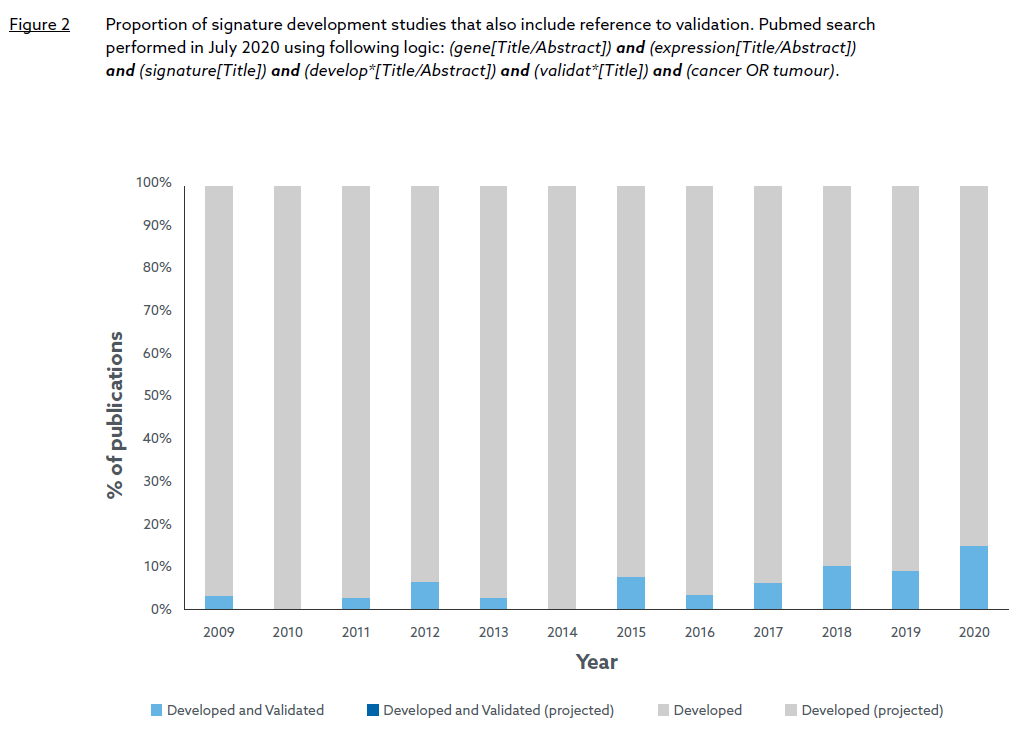
Principally, they noted that many early gene signatures were developed on small training sets (increasing the risk of over-fitting), and lacked external validation resulting in low reproducibility in independent datasets. A Pubmed search confirms that signature validation was rarely performed in the microarray-led growth period with no publication explicitly referring to “validation” in the title between 2003 and 2008, and only six publications between 2009 and 2014 (Figure 2). However, recent trends suggest that the situation has slowly improved in the RNASeq-led growth phase, culminating this year with over 15% of developed signatures undergoing some form of validation. Whether this trend translates to a higher proportion of FDA approved signatures in the future remains to be seen but it does suggest that lessons have been learned and more rigorous practises are now being applied to RNA signature development.
Two further issues continue to impede progress to the clinic. Firstly, for many signatures, the gain in predictive accuracy compared to more established prognostic factors better suited to clinical testing is either insufficient or unquantified. Indeed, the PAM50 signature, which enables classification of breast cancer into four prognostic subtypes4, is justifiably regarded as clinically influential (and now forms the basis of the Prosigna assay), but at the time was not immediately adopted in the clinic because cheaper and more efficient surrogates such as immunohistochemical measurement of hormone receptor (HR) and HER2 status performed equally well. Secondly, some signatures are cohortdependent where individual sample scores rely on information from other samples in the same cohort. This results in unstable and non-reproducible scores that cannot be validated for use in prospective clinical testing where samples are measured one at a time.
Almac signature discovery and validation process
Almac Diagnostic Services has long been a strong advocate for the use of robust best practices and standards in signature development and validation, exemplified by our active contribution to the MAQC initiative in 20101. Based on this experience, we have an established bioinformatics Biomarker Discovery process applicable to both cDNA microarray and RNASeq platforms designed to meet MAQC standards and avoid the common pitfalls highlighted above.
The initial phase of the process consists of a series of data QC steps, which include Almac’s proprietary Exploratory Analysis (EA) tool to identify and reduce any technical effects that may confound signature generation.
This is followed by the signature discovery/ development phase, which begins with feature selection and performance metric generation carried out under cross-validation, and then application of a machine learning method appropriate to the endpoint (whether discrete or continuous). Multiple factors guide final model selection including statistical performance, biological relevance and independence from established clinical biomarkers. The chosen model is always further validated using independent test data.
As a result of this process, Almac Diagnostic Services has discovered and validated several proprietary biomarkers such as our own DNA Damage Immune Response (DDIR), Angiogenesis, Epithelial-Mesenchymal Transition (EMT), ProstateDx and ColDx signatures. Almac’s ColDx and DDIR signatures, originally developed on microarray platforms, have been independently clinically validated by the Cancer and Leukemia Group B (CALGB) and SWOG consortiums respectively5, 6. Furthermore, the DDIR signature has recently been transferred to the Illumina RNA Exome platform, undergoing a rigorous analytical validation process that meets both Clinical Laboratory Improvement Amendments (CLIA) and Clinical and Laboratory Standards Institute (CLSI) guidelines7. To the author’s knowledge, this is one of the first analytical validation studies of a gene expression signature on RNA-Seq technology.
The wisdom of crowds: Almac claraT Total mRNA report
Recently we have seen an increased interest in companion diagnostic gene expression signatures from our Pharmaceutical partners. This is perhaps because the complexity of tumour biology underpinning the response to certain therapies is not adequately captured by immunohistochemical or DNA analysis. For example, the response to immune targeted therapies may be determined by a complex interaction between tumour and stromal molecular pathways which are often dysregulated at a gene expression level through mechanisms other than DNA mutation.
This has led Almac Diagnostic Services to develop claraT, a unique software-driven solution that integrates a diverse set of pan-cancer gene expression signatures into a comprehensive, easy-to-interpret cohort report. Over 90 signatures representing all 10 Hallmarks of Cancer8 are included in the report with each signature selected for inclusion based on a set of rigorous scientific and technical criteria including:
- literature-based review of scientific and clinical rationale.
- level of validation and clinical utility.
- feasibility of implementing published signature methodology.
The integrative approach compensates for any potential shortcomings at the individual signature level by use of information from other signatures, thus increasing the likelihood of discovering accurate and clinically relevant disease subtypes. It also allows efficient visualisation of the key discriminating biologies within either a large cohort or an individual tumour sample. The full end-to-end solution from raw sequence data to claraT report takes a matter of hours regardless of cohort size, saving researchers months of effort in selecting and implementing a similar number of signatures themselves. Thus, claraT accelerates the interpretation of complex datasets, extracting value from gene expression markers even for those without specialist computational knowledge.
Closing remarks
Whilst the potential of gene expression signatures remains largely unfulfilled, the increased drive in recent years to meet the standards first advocated by MAQC a decade ago provides hope that signatures can begin to progress more frequently beyond the development phase and translate to patient benefit.
Almac Diagnostic Services will continue to look to the future and support these efforts by promoting robust signature development and best practice, whilst drawing on our experience to offer claraT, a powerful computational tool that distils some of the most prominent cancer gene signatures to emerge over the last 25 years into a single reporting solution.
More information
Almac Diagnostic Services has recorded a webinar on the analytical validation of gene expression signatures, presented by Dr Katarina Wikstrom, entitled “Overcoming the challenges of taking RNA biomarkers into clinical trials.”
Download this complimentary webinar to explore key considerations in the development & analytical validation of developing RNA biomarkers suitable for clinical stratification.
References











 Professor Richard Kennedy
Professor Richard Kennedy